国家药品监督管理局药品审评中心(CDE)发布了关于微生物限度检查的申报资料要求(征求意见稿),该文件旨在进一步规范药品研发与注册过程中的微生物质量控制,保障药品安全。本文将对该要求进行详细解读,为相关从业者提供清晰的指导。
一、法规背景与核心目标
此次发布的征求意见稿,是CDE在《中华人民共和国药品管理法》、《药品注册管理办法》以及《中国药典》通则<1105>、<1106>等现有法规框架下的进一步细化。其核心目标在于:
- 统一标准:明确和统一药品注册申报中微生物限度研究资料的技术要求,减少审评过程中的不确定性。
- 强化控制:引导申请人建立科学、全面的微生物质量控制体系,从源头保障非无菌药品的微生物安全性。
- 基于风险:强调基于产品特性、生产工艺和预期用途进行风险评估,实施针对性的微生物控制策略。
二、申报资料要求的关键内容解读
申报资料需系统性地呈现微生物限度控制的各个方面,主要涵盖以下几点:
- 微生物限度标准制定依据:
- 必须详细说明各品种微生物限度标准(包括需氧菌总数、霉菌和酵母菌总数、控制菌检查)的制定依据。
- 应参考《中国药典》、ICH Q4B、USP等国内外权威药典,并结合原料来源、生产工艺、处方组成、给药途径及临床应用风险进行综合论证。对于标准中收载的品种,若拟订标准与药典不同,必须提供充分的验证和合理性论证数据。
- 分析方法开发与验证:
- 方法适应性:必须提供详细的微生物计数方法和控制菌检查方法的开发资料,证明所选方法适用于待检样品。重点需关注产品的抑菌性,通过方法适用性试验(回收率试验)证明方法的有效性。
- 验证资料:需提供完整的验证方案和报告,验证内容应涵盖方法的专属性、准确性(回收率)和重现性等关键指标。对于薄膜过滤法、稀释法或中和法等特殊处理,需详细说明其必要性和有效性。
- 检测结果与数据汇总:
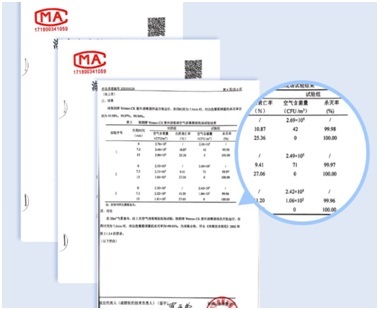
- 申报时应提供代表性批次(如工艺验证批次、稳定性研究批次)的微生物限度检查结果。数据应真实、完整、可追溯。
- 对于检测中发现的任何微生物,特别是检出控制菌或微生物计数超警戒限/行动限的情况,需进行分析调查,评估其对产品质量和患者安全的风险,并说明已采取的纠正预防措施。
- 稳定性研究中的微生物考量:
- 微生物限度检查是稳定性研究的重要组成部分。申报资料需说明在稳定性考察方案中微生物检查的测试频率和考察点。
- 通过稳定性数据,论证产品在拟定的贮藏条件下,微生物质量属性能够始终符合标准要求,从而支持产品的有效期和贮藏条件。
- 生产工艺的微生物控制:
- 资料应体现“质量源于设计”的理念,阐述生产工艺中如何控制微生物污染,例如原料料的质量标准、生产环境(如D级洁净区)、设备清洁消毒、中间产品储存条件与时限等。
- 提供相关验证资料,如清洁验证、保存时限验证中涉及的微生物数据。
三、对企业的实践建议与挑战
- 尽早规划,融入研发全流程:微生物限度控制策略不应是注册申报前的“补课”,而应尽早纳入药品研发阶段。从处方设计、工艺开发到包装选择,均需同步考虑微生物控制因素。
- 加强方法学研究:方法是基础。企业需投入资源进行扎实的方法开发与验证,尤其对于中药、益生菌、含防腐剂或具有抑菌性的产品,其方法适用性试验至关重要。
- 建立系统的微生物监控体系:不局限于最终产品的检验,应建立覆盖原料、辅料、包装材料、生产环境、中间产品及成品的完整微生物监控体系,并基于数据趋势进行风险管理。
- 注重调查与数据完整性:对任何异常结果(OOS/OOT)进行彻底调查是法规的基本要求。所有微生物检测数据必须保证真实、准确、完整、可追溯,符合数据完整性(ALCOA+原则)要求。
- 挑战与应对:对于复杂制剂(如微生态制剂)、新型给药系统或来源于动植物的原料,其微生物限度方法的建立和标准制定可能面临更大挑战。建议加强与CDE的早期沟通,寻求前瞻性科学建议。
###
CDE此次发布的微生物限度申报资料要求(征求意见稿),标志着我国药品微生物质量控制监管向更科学、更严谨、更与国际接轨的方向迈进。对企业而言,这既是提升自身质量管理水平的契机,也提出了更高的技术要求。深入理解法规精神,系统构建从研发到生产的微生物控制体系,是保证药品安全有效、成功通过注册审评的必由之路。相关单位应积极研究该征求意见稿,并及时反馈意见,共同完善这一重要技术指南。